こんにちは!
それでは今回も化学のお話やっていきます。
今回のテーマはこちら!
動画はこちら↓

動画で使ったシートはこちら(¹³C-NMR)
それでは内容に入っていきます!
¹³C-NMRを測定する意義
まず始めに、なぜ¹H-NMRだけでなく¹³C-NMRを測定するのかということからお話ししていきます。
結論を先に言うと、¹³C-NMRのメリットは¹H-NMRの複雑さを回避できることにあります。
¹H-NMRではスピン-スピン分裂が顕著に現れるため、非等価な隣接水素が何種類もあると、何本に分かれているのかもよくわからないような多重線が得られることがあります。


こうなると、もはや人の手では解析できず、コンピュータに頼らざるをえない状態になります。
¹³C-NMRではこの複雑さが消すことができるというのです。
では、その理由を見ていきましょう。
NMR活性な核種の天然存在比
なぜ炭素原子を使うのか
有機化合物に含まれる元素の種類は少なく、炭素、水素のほか酸素や窒素、ハロゲンなどがあります。

この中で、炭素と水素以外の元素はヘテロ原子と呼ばれ、この原子の周りは反応性が高くなるなど、その化合物の物性を大きく決めうる要素となります。
しかし、ヘテロ原子をもたない有機化合物も多く存在しており、吸収ピークが観測されない可能性があります。
したがって、未知の有機化合物の構造をNMRによって調べるためには、ほとんどの有機化合物がもっている炭素か水素のみが適していることになります。
NMR活性と天然存在比
そこで次にNMR活性、すなわち\(\displaystyle 0\)ではない核スピンをもつ同位体について考えます。
水素の場合は質量数\(\displaystyle 1\)の水素原子、炭素の場合は質量数\(\displaystyle 13\)の炭素原子になります。
ここで注目すべきはそれぞれの天然存在比です。
水素の場合実に\(\displaystyle 99\%\)以上の原子が質量数\(\displaystyle 1\)であるのに対し、炭素の場合はほとんどが質量数\(\displaystyle 12\)であり、\(\displaystyle 13\)の炭素原子は約\(\displaystyle 1.1\%\)しか存在していません。
天然存在比が低いということは、同一の分子内でカップリングするほど近くに位置する可能性がとても低いということなので、炭素原子同士のカップリングは起こらず、¹H-NMRに比べてスペクトルが単純化しやすいことになります。
¹³C-NMRと¹H-NMRの違い
ここで¹、³C-NMRと¹H-NMRの違いを2つ見ていきます。
ピーク強度
1つ目はピークの強度が大きく異なるということです。
¹³Cは天然に少ししか存在していないので、ピークが見にくいというのはイメージしやすいかと思います。
ちなみに¹H-NMRにおいてH-¹³Cカップリングが見えないのはこの天然存在比が理由です。
ただ、原子1個当たりの共鳴強度で比べたとしても、¹Hの方がピークが大きくなるため、二重の効果で¹³C-NMRのピークは小さくなります。
仮に同じ測定条件で両方のピークをとったとすると、¹³Cのピークは¹Hの約6000分の1になるそうです。
これを精度よく測定するためには、長時間測定するというのもいいのですが、もっと現実的な方法があります。
それがフーリエ変換を使って数学的に解析するということです。
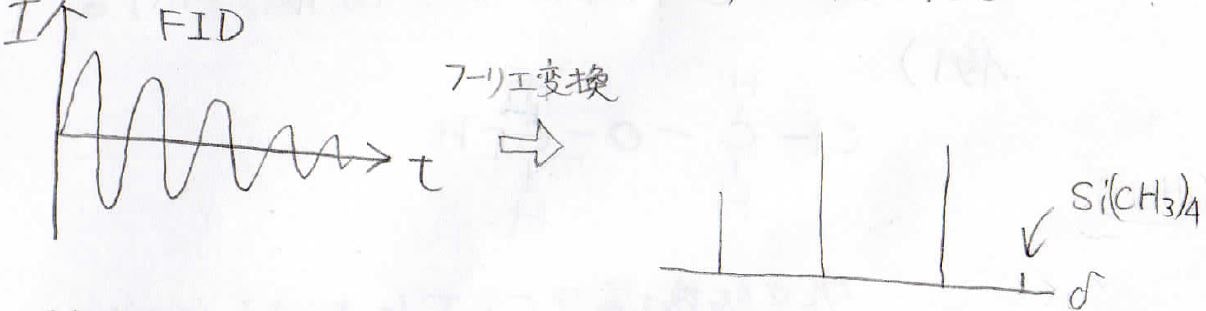
FTというのがFourier Transform、すなわちフーリエ変換を指すのですが、得られる信号をいくつかの周波数のラジオ波パルスを足し合わせたものと考えることで、短時間の測定でも大きなピークが得られることになります。
この測定原理については過去に挙げたNMRの原理という記事でもお話ししているので、もしよければそちらも見てください。

化学シフトの領域
そして2つ目の違いがスペクトルが現れる化学シフトの領域です。
¹Hの場合、\(\displaystyle 10\ \rm{ppm}\)くらいまででほとんどのピークをとらえることができますが、¹³Cの場合は約\(\displaystyle 200\ \rm{ppm}\)までいくことがあり、広い範囲にわたるラジオ波の吸収が起こっています。
¹³C-¹Hカップリング
では今度、¹³C-¹Hでのカップリングというものを紹介します。
ここまで¹³C-NMRは単純なスペクトルを得られますと言ってきたのですが、¹³Cの近くに¹³Cはなくても¹Hがあることがほとんどです。
実際、これらは大きなカップリング定数を持つため、例えば、ブロモエタンの¹³C-NMRはこのようになります。
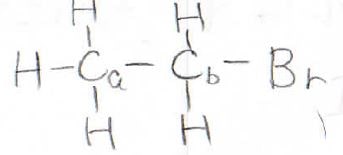
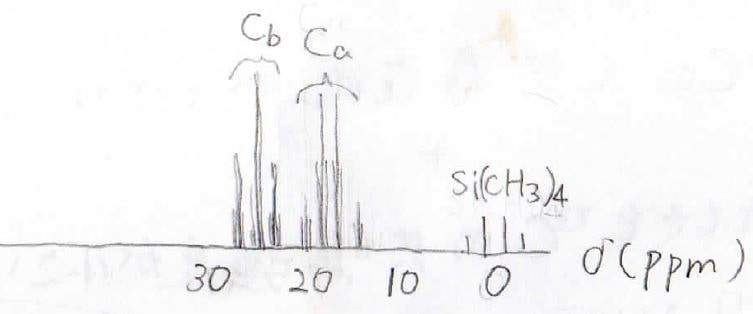
そのカップリング定数は\(\displaystyle 125\sim 200\ \rm{Hz}\)にまでのぼります。
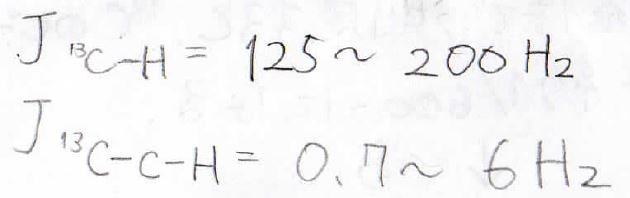
しかし、隣接している炭素原子に結合している¹Hとのカップリング定数は\(\displaystyle 0.7\sim 6\ \rm{Hz}\)と、一気に小さくなります。
このままだと、せっかく単純なスペクトルを得るために¹³C-NMRを測定したのに、そのメリットがありません。
では、結局意味がないのかというと、そうではありません。
実は、¹³C-¹Hカップリングは広帯域水素デカップリング、もしくはプロトンデカップリングという手法で取り除くことができます。
その方法は、次のようなイメージです。
例えば、下の物質に対して外部磁場\(\displaystyle 7.05\ \rm{T}\)で低分解能のNMRの測定をしたときには、炭素原子が\(\displaystyle 73\ \rm{Hz}\)、水素原子が\(\displaystyle 300\ \rm{Hz}\)の電磁波を吸収します。

ここで両方の電磁波を同時に照射すると、炭素と水素が同時に共鳴することになります。
そして、スピン状態が\(\displaystyle \alpha\)から\(\displaystyle \beta\)へ交換する速さは、炭素よりも水素のほうがとても速いため、炭素から見たときには水素の動きが追えなくなります。
こうなると、NMRの共鳴ピークははっきりと分かれなくなり、デカップリングします。
結果、水素と結合している炭素のピークでも単一線として現れることになります。
ブロモエタンのスペクトルはこうなります。
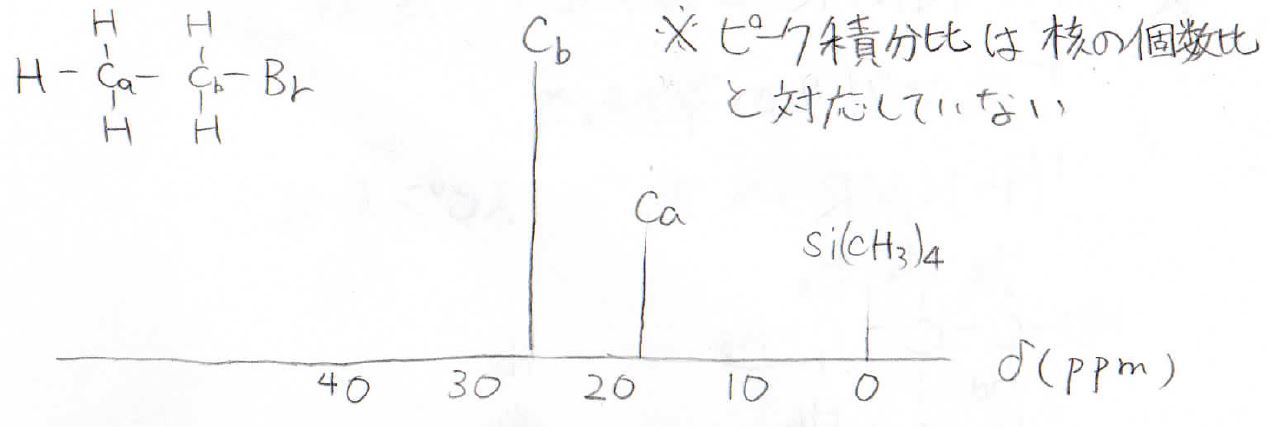
ここで、注意してもらいたい点が一点あって、¹H-NMRではピークの積分比がそのまま個数比でしたが、プロトンデカップリングさせた¹³C-NMRでは個数比と対応しません。
ぜひ知っておいてください。
代表的な構造に対応する化学シフト値
そして、この単純なスペクトルを使って化合物の同定ができます。
ここに示すのは、代表的な構造の化学シフトです。
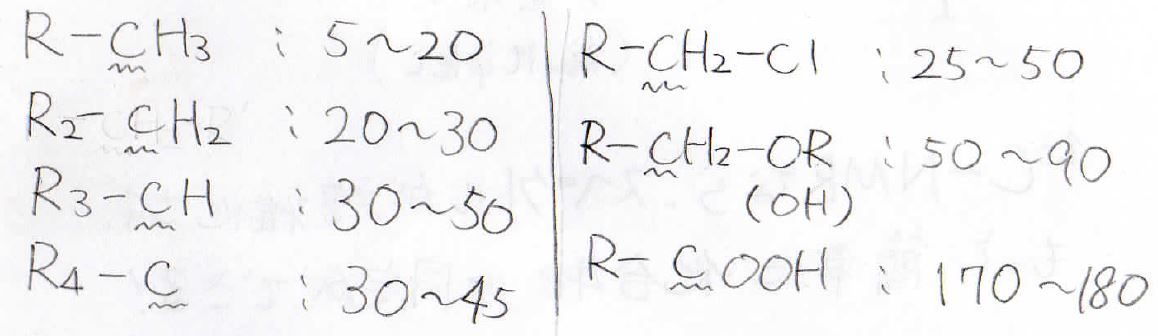
有機化学の教科書なら必ず載っているはずなので、このほかの構造についてはご自身の教科書を見てみてください。
異性体を持つ化合物の同定
¹³C-NMRでは等価な炭素原子の種類の数だけピークが現れるため、異性体が存在する化合物の同定に特に真価を発揮します。
例えば、元素分析、質量分析にて未知物質の分子式が\(\displaystyle \rm{C_7H_{14}}\)とわかっているとします。
しかし、\(\displaystyle \rm{C_7H_{14}}\)にはこのようなシクロアルカンの異性体があります。
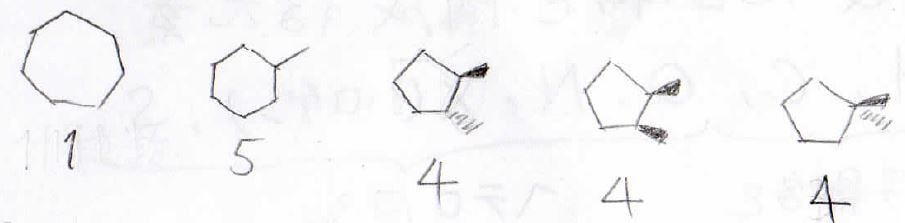
このほかにもありますし、何ならアルケンかもしれません。
こういった場合に注目してほしいのが等価な炭素原子の個数です。
構造の下に示した数字は、等価な炭素原子の数を表しています。
この数字と¹³C-NMRのピークの本数が異なる場合は、その構造の可能性を否定することができます。
練習問題
ということで、最後に練習問題をやってみましょう。
分子量が\(\displaystyle 70\)の炭化水素、すなわち構成する元素が炭素と水素のみである化合物で¹³C-NMRのピークが4本ありました。
考えられる構造をすべて答えてください。
不飽和度は\(\displaystyle 1\)であり、シクロアルカンかアルケンだと考えられます。
あとは考えられる異性体の中から化学的に等価な水素原子が4個のものを探すと、メチルシクロブタンと3-メチル-1-ブテンが該当します。
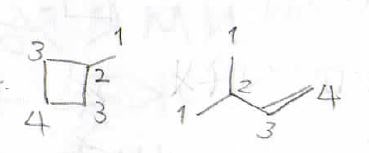
あとは付加反応が起こるか調べれば、どちらの化合物か断定することができます。
このように、¹³C-NMRでは化学シフトの値を無視して、ピークの本数だけでも化合物がわかってしまうことがありますので、知っておいてください。
まとめ
はい、今回の内容は以上です。
間違いの指摘、リクエスト、質問等あれば、Twitter(https://twitter.com/bakeneko_chem)かお問い合わせフォームよりコメントしてくださると、助かります。
それではどうもありがとうございました!