こんにちは!
それでは今日も化学のお話やっていきます。
今回のテーマはこちら!
動画はこちら↓


動画で使ったシートはこちら(Friedel Crafts)
求電子置換反応の一般式
まず、今回お話しする求電子置換反応とは、どういうものなのかを一般式を用いて、説明します。
前回の記事で、ベンゼン環の平面に対して垂直方向には、\(\displaystyle \pi\)電子雲が形成されていると言いました。

ベンゼンそのものは熱力学的にきわめて安定な物質ですが、一定の条件のもとでは、その高い電子密度によって、求電子剤と反応する可能性があります。
しかし、アルケンと求電子剤との付加反応とは異なり、ベンゼン環が非常に安定であるため、置換反応が優勢となります。
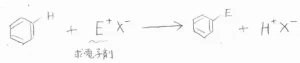
求電子置換反応の機構は、まず求電子剤がベンゼン環へ求電子的に攻撃します。
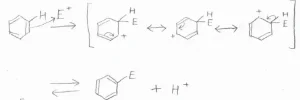
そうしてできたカチオン中間体は、式のような共鳴構造をとり安定となります。
ここから、プロトンが脱離することで、ベンゼン環が復活するという流れです。
ポテンシャル図で書くと、こちらのようになります。
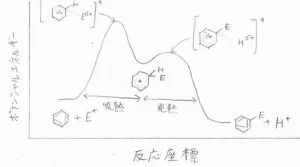
求電子剤が攻撃してカチオンになる素反応では、ベンゼン環がなくなるので、熱力学的には不安定となります。
ここからプロトンが脱離してベンゼン環が復活する素反応で、大きく安定化します。
つまり、1段階目は吸熱反応で、2段階目は発熱反応となります。
活性化エネルギーは、1段階目のほうが大きいため、こちらが律速段階となります。
ここからは、実際の反応を例にお話ししていきます。
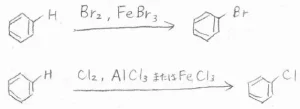
ハロゲン化
まず、ハロゲン化についてですが、塩素と臭素はそこまで求電子的ではないため、Lewis酸触媒による活性化が必要になります。
フッ素との反応は、非常に発熱的で、触媒がなくとも爆発的に進行するため、制御ができません。
ヨウ素との反応は、全体で吸熱的となるため、ほとんど進行しません。
これら2つについては、別の合成法で作るのが一般的です。
臭素への置換反応の機構は、こちらに示したとおりです。
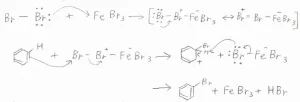
まず、臭素分子の孤立電子対がLewis酸触媒へ移動して、錯体を形成します。
これにより臭素-臭素間の結合が分極して、末端の臭素原子が求電子的になります。
この臭素原子がベンゼンへ求電子攻撃すると同時に、\(\displaystyle \rm{FeBr}\)\(_4^-\)が安定な脱離基として脱離することで、カチオン中間体ができます。
そして、今度は\(\displaystyle \rm{FeBr}\)\(_4^-\)が塩基として作用することで、カチオン中間体からプロトンが引き抜かれて、ブロモベンゼンが生成するという流れです。
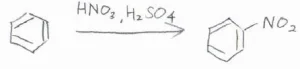
ニトロ化
続いて、ベンゼンのニトロ化について、お話しします。
濃硝酸はそこまで求電子的ではないので、ベンゼンと混ぜるだけではニトロ化はあまり起こりません。
そこまで高くない温度でもニトロ化を起こすために、濃硫酸で活性化させる必要があります。
これが、ニトロ化の反応機構です。
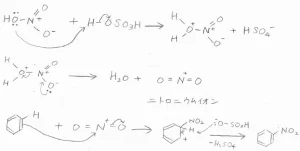
まず、硝酸分子の酸素原子がプロトン化されます。
それから、優れた脱離基として水が脱離すると同時にニトロニウムイオン\(\displaystyle \rm{NO}\)\(_2^+\)が生成します。
これが求電子剤としてベンゼンと反応することで、ニトロベンゼンが生成します。
スルホン化
次に、スルホン化についてお話しします。
濃硫酸\(\displaystyle \rm{H_2SO_4}\)\(\)もそこまで求電子的ではないので、そのままではベンゼンとの反応はあまり進行しません。
そこで、より活性の高い発煙硫酸という試薬を使います。
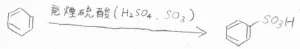
発煙硫酸とは、濃硫酸に\(\displaystyle 8\%\)の三酸化硫黄\(\displaystyle \rm{SO}\)\(_3\)を加えた混合物です。
3つの酸素原子から電子求引されることで、中心の硫黄分子が十分に求電子的となっているため、ベンゼンを求電子攻撃し、ベンゼンスルホン酸が得られます。
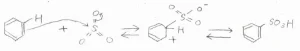
ただし、この反応は逆反応も起こります。

これは、三酸化硫黄が熱力学的に不安定で、水和して濃硫酸となったときに、非常に大きな熱を発するためです。
ベンゼンスルホン酸を希硫酸中で加熱すると、スルホン基が水素に置換されます。
これを利用すると、ベンゼン誘導体の配向性をコントロールするための保護基として、スルホン基を利用することができます。
また、ほかの利用法として、合成洗剤があります。
大きなアルキル基が付いたベンゼン誘導体をスルホン化した後、水酸化ナトリウムを加えると、ナトリウム塩となります。
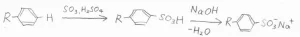
分子内に疎水的な部分と親水的な部分がどちらもあるので、これは界面活性剤としてはたらきます。
ただし、こういった物質は生物分解されにくいため、現在は使用頻度が減ってきています。
また、別の用途としては、有機反応によく用いられる塩化スルホニルの合成があります。
ベンゼンスルホン酸ナトリウムに五塩化リンまたは塩化チオニルを反応させると、塩化ベンゼンスルホニルが得られます。
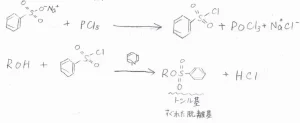
これをアルコールと反応させれば、脱離能の小さなヒドロキシ基を脱離能の大きなトシル基に変換できて、求核置換反応が起こせるようになります。
また、塩化スルホニルはスルホンアミドの前駆体にもなります。
アニリンから活性を落としたアセトアニリドをスルホン化した後、塩化スルホニルにして、そこにアンモニア水もしくはアミンを反応させると、スルホンアミドになります。
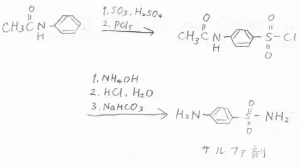
これを加水分解して塩酸塩とした後、塩基で中和させると、合成抗菌剤や化学療法剤であるサルファ剤を得ることができます。
サルファ剤はp-アミノ安息香酸と類似の構造をもっていることで、葉酸の合成を阻害し、バクテリアの細胞を死滅させるはたらきをします。
現在は、新しい抗生物質や耐性菌の誕生に伴い、単体で使用される機会は減っていますが、医薬品化学の発展に寄与した重大な発見として知られています。
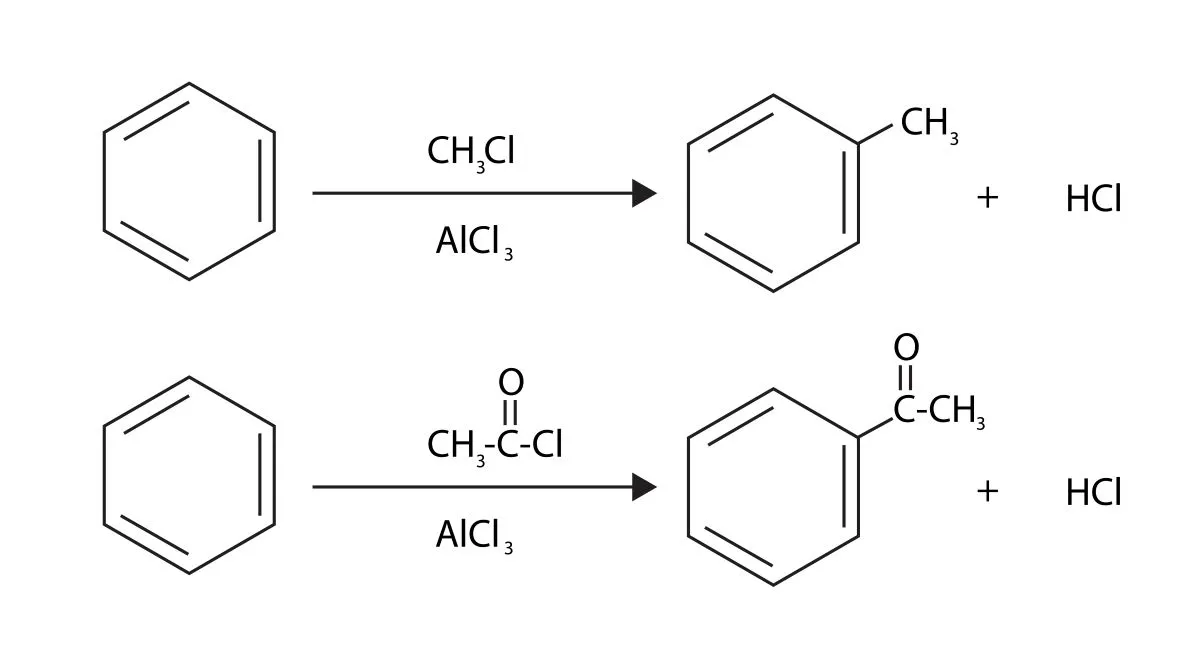
Friedel-Craftsアルキル化
続いて、こちらは求電子置換反応によるベンゼンのアルキル化の一般式です。
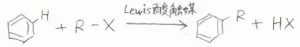
ハロアルカンをLewis酸触媒で活性化することで、求電子剤としてベンゼンと反応が起こります。
この反応は、Friedel-Craftsアルキル化と呼ばれるものです。
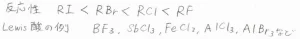
ハロアルカンの反応性は、ヨードアルカンが最も低く、フルオロアルカンが最も高くなります。
Lewis酸触媒の例としては、3価の塩化鉄、塩化アルミニウム、臭化アルミニウムなどが使われます。
Friedel-Craftsアルキル化の例として、下記の反応について、反応機構を考えてみます。
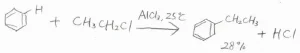
まず、クロロエタンの塩素原子上にある孤立電子対が移動して、塩化アルミニウムと錯体を形成します。
これにより塩素原子が結合している炭素原子周辺の電子密度が小さくなり、求電子的になります。
これがベンゼンに求電子攻撃すると同時に、\(\displaystyle \rm{AlCl}\)\(_4^-\)が脱離します。
また、これが塩基として、カチオンからプロトンを引き抜くことで、エチルベンゼンが生成します。
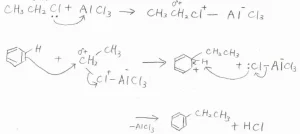
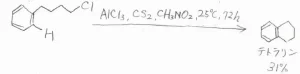
この反応は、分子内でも起こることがあって、例えば、4-クロロブチルベンゼンにLewis酸触媒を加えて長時間待つと、テトラリンが得られます。
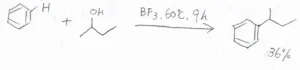
また、この反応は必ずしもハロアルカンである必要はなく、カルボカチオンの前駆体であれば進行します。
ここには、アルコールを用いた場合とアルケンを用いた場合の例を示しています。
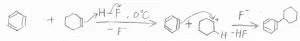
逆に、ハロゲン化物であったとしても、ハロアルケンやハロアレーンなどハロゲン化物イオンがとれてできるカチオンが生成不可能なほど不安定な場合は、Friedel-Craftsアルキル化は進行しません。
Friedel-Craftsアルキル化の欠点
そして、実は、この反応には2つ大きな欠点があることが知られています。
多アルキル化
まず、アルキル基が電子供与性であるために、ベンゼン環内の電子密度が増大することで、さらに求電子置換が起こりやすくなるというものです。
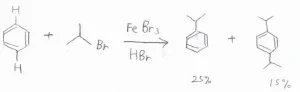
これにより、一置換体だけを作りたいのに、二置換体もたくさんできてしまったということが起こりやすいです。
この現象は、前回紹介したようなハロゲン化、ニトロ化、スルホン化では見られません。
これは、これらの置換基が電子求引性であるために、ベンゼン環内の電子密度を減少させて、求電子置換が起こりにくくするためです。
転位
そして、Friedel-Craftsアルキル化の2つ目の欠点は、カルボカチオンの転位が起こる可能性があるということです。
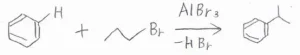
例えば、1-ブロモプロパンを使ったのに、イソプロピルベンゼンが生成するといったことが起こります。
これは、Lewis酸存在下でこのようなヒドリド移動が起こるためです。
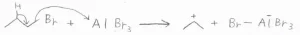
以上のことから、Friedel-Craftsアルキル化で目的の物質だけを高い収率で得ることは非常に困難で、合成化学で用いられることはほとんどありません。
Friedel-Crafts アシル化
実験室で、求電子置換反応でベンゼンと新たな炭素-炭素結合を作りたいときには、ハロアルカンの代わりにハロゲン化アシルが使われます。

アシル基は電子求引性で、ベンゼン環内の電子密度を減少させるため、さらなる求電子置換はほとんど起こりません。
また、転位が起こる可能性もありません。
反応剤である塩化アシルは、カルボン酸と塩化チオニルの反応によって得られます。

酸素原子上に孤立電子対があるので、これがLewis塩基としてはたらくと、Lewis酸触媒と結合して共鳴安定化した錯体を形成できます。
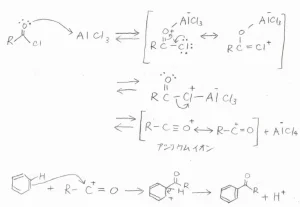
この錯体は、アルミニウムが塩素原子と結合した構造異性体と化学平衡にあり、そこから\(\displaystyle \rm{AlCl}\)\(_4^-\)が脱離することで、求電子剤であるアリシウムイオンが生成します。
これがベンゼンに求電子攻撃することで、1-フェニルアルカノンが生成します。
ただし、この生成物もLewis酸と錯体を形成するため、水を用いた後処理が必要になります。
アシル化は、酸無水物を用いた方法もあります。
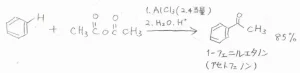
中央の酸素原子とLewis酸で結合を作った後、アリシウムイオンができるという反応機構です。
アルキルベンゼンへの還元方法
そして、このアシル基はいくつかの方法によって、アルキル基まで還元することができます。
パラジウム触媒共存下の水素化
下の反応式は、その一例です。
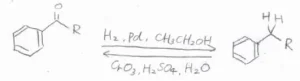
アルキル基をアシル基に戻したい場合は、酸性溶液中で酸化剤である酸化クロム(III)と反応させます。
アルキル基へ還元する方法として、ここではあと4つ紹介します。
ヒドリド還元剤を用いた水素化
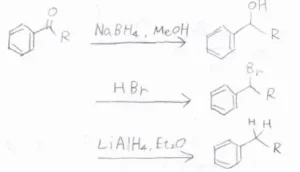
水素化ホウ素ナトリウムでケトンをアルコールまで還元した後、臭化水素を加えて求核置換反応させます。
これに水素化アルミニウムリチウムを加えることで、臭素原子が水素原子に置換されて、アルキル基となります。
Clemmensen還元
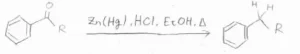
これは、反応機構は完全には解明されていませんが、濃塩酸中で亜鉛アマルガムと一緒に加熱還流させることで、アルキル基まで還元することができます。
酸性条件であるため、酸に弱い基質に対しては行うことができません。
Wolf-Kishner還元
塩基性条件で還元する方法としては、Wolf-Kishner還元があります。
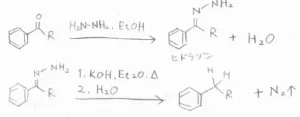
まず、カルボニルとヒドラジンを反応させてヒドラゾンとします。
高温条件下でヒドラゾンを塩基と反応させると、窒素ガスを発生させながら分解し、炭化水素が生成します。
チオアセタールの脱硫還元
また、中性条件で行う反応として、チオアセタールの脱硫還元があります。
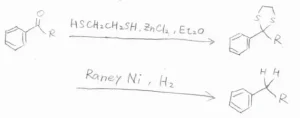
カルボニル基は、1,2-エタンジチオールと反応して、環状チオアセタールを生成します。
これをRaneyニッケル触媒存在下で水素と反応させると、脱硫することができて、カルボニル基はメチレン基へ変換されます。
Raneyニッケルとは、ニッケルとアルミニウムの合金から水酸化ナトリウム水溶液によりアルミニウムだけを溶解除去してできる、穴だらけでスポンジ状になったニッケルのことです。
表面積が大きいため、触媒活性が高くなります。
Gattermann-Koch反応(ホルミル化)
最後に、ベンゼンのホルミル化の方法についてお話しして終わります。
最も小さな塩化アシルは塩化ホルミルですが、熱力学的にとても不安定であるため、すぐに一酸化炭素と塩化水素に分解してしまい、Friedel-Craftsホルミル化の反応剤として直接使うことはできません。
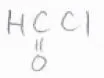
その代わりの方法として、Gattermann-Koch反応というものがあります。
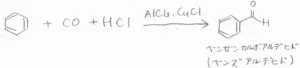
塩化水素とLewis酸触媒の存在下で、ベンゼンと一酸化炭素を混合し、圧力をかけることで、ベンズアルデヒドを得ることができます。
求電子剤が生成する機構は、下記のとおりです。
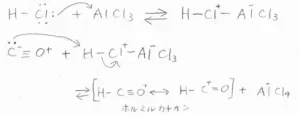
まず、塩化水素がLewis塩基として、Lewis酸触媒と錯体を形成します。
ここから、一酸化炭素の炭素原子がプロトンを引き抜き、ホルミルカチオンが生成します。
これが求電子剤としてベンゼンと反応することになります。
ホルミルカチオンはアセチレンの酸素類縁体とみなすことができて、¹³C-NMRやIRによって、その存在も確認されています。
まとめ
今回の内容は以上です。
間違いの指摘、リクエスト、質問等あれば、X(https://X.com/bakeneko_chem)かお問い合わせフォームよりコメントしてくださると、助かります。
それではどうもありがとうございました!