こんにちは!
それでは今日も、化学のお話をしていきます。
今回のテーマはこちら!
動画はこちら↓

動画で使ったシートはこちら(orientation、orientation 2)
配向性とは
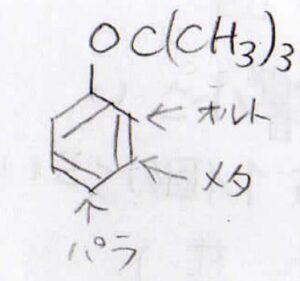
例えば、ベンゼンの一置換体があったとします。
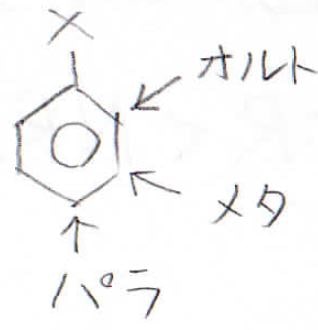
これにもう1つ置換基をつけようとしたときに、どの水素原子が置換されやすいかという傾向のことを配向性と言います。
Xと隣接したものはオルト置換体、その隣を置換したものはメタ置換体、正反対の位置を置換したものはパラ置換体と呼びます。
配向性を決定づける要因
ではここから配向性を決定させる要因についてお話ししていきます。
ベンゼンは、そもそも結合角ひずみが無く共鳴効果もあるため、とても安定なのですが、\(\displaystyle \pi\)電子が6個もあるので、環の電子密度が大きく、求電子的な化学種と反応することがあります。
置換基がベンゼン環中の電子密度が変化させることで、反応性も変化します。
電子密度が変化する要因は、誘起効果と共鳴効果の2つです。
誘起効果
まず、誘起効果はI効果とも呼ばれ、電気陰性度の差が主な原因となってベンゼン環内の電子密度が変化します。
この効果は\(\displaystyle \sigma\)結合の中で現れるものなので、遠くの結合にはあまり影響がありません。
電子密度を上げる置換基は電子供与基またはドナー基と呼ばれ、電子密度を下げる置換基は電子受容基またはアクセプター基と呼ばれます。
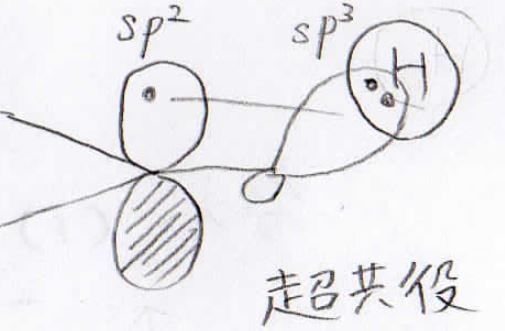
代表的な電子供与基は、アルキル基です。
電気陰性度の差はありませんが、超共役によりベンゼン環内の電子密度が大きくなります。
電子求引基は、有機化学でよく出てくる置換基の中では、アルキル基以外と考えて問題ありません。
ハロゲンやヒドロキシ基、ニトロ基、アミノ基、スルホン基などが該当します。
共鳴効果
続いて、もう一方の要因となる共鳴効果についてお話しします。
これはR効果とも呼ばれ、それぞれ共鳴構造に起因しています。
\(\displaystyle \pi\)結合を介しているので、遠くの結合にも関与することができます。
例えば、置換基に孤立電子対があった場合、このような共鳴構造をとることができるので、ベンゼン環内の電子密度は増大することになります。
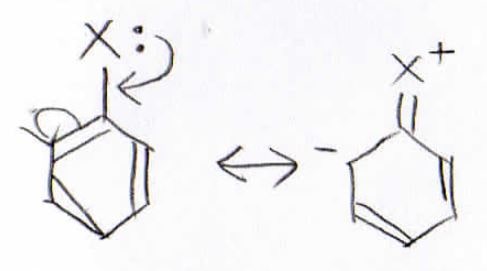
そのため、ハロゲンやアルコキシ基はベンゼン環の電子密度を増大させます。
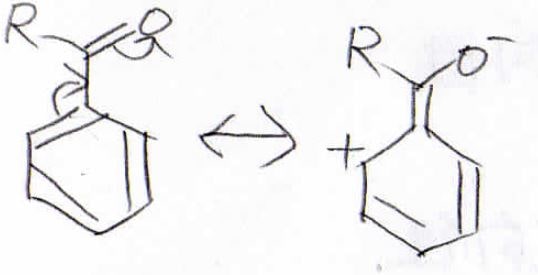
反対に、ニトロ基やアシル基は電子密度を小さくします。
これらは、酸素原子上に負電荷を抱え込める共鳴構造を書くことができるため、電子密度は小さくなります。
求電子剤とベンゼン置換体の反応
では実際に、ベンゼンの一置換体に求電子剤を反応させるとどうなるか考えていきましょう。
誘起効果
電子供与基
まず、置換基がアルキル基だった場合、この置換体はこのような共鳴構造が安定となるのでオルト位とパラ位の電子密度が増大することになります。
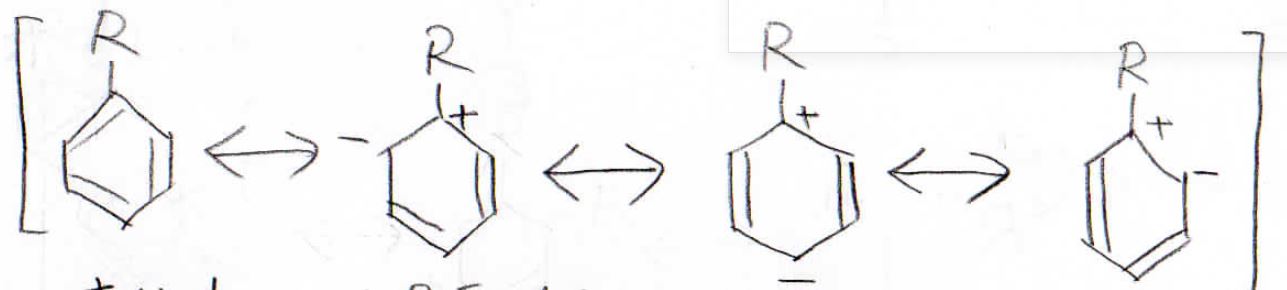
そのため、ここが求電子剤と反応しやすい位置だという事になります。
電子受容基
続いて、置換基がブロモ基だった場合を考えます。
ブロモ基には孤立電子対があるため、共鳴効果もありますが、ここでは誘起効果だけを考えた場合を示します。
臭素と結合している炭素上に負電荷がある共鳴構造が安定となるので、このような共鳴構造を書くことができます。
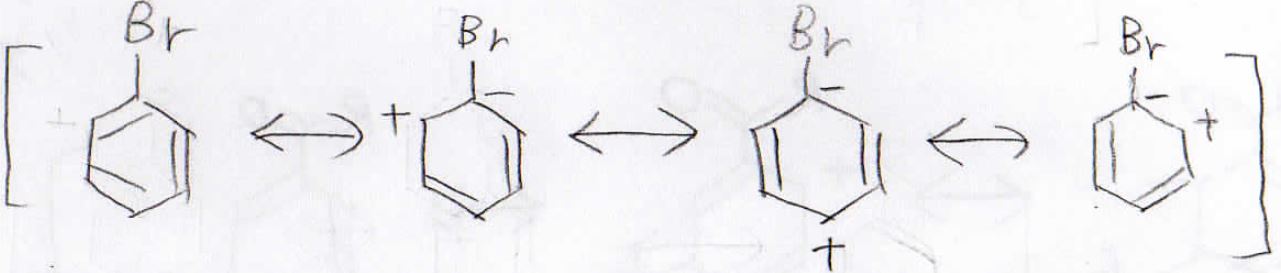
すると、このようにオルト位とパラ位の電子密度が減少し、相対的にメタ位の反応性が上がることになります。
共鳴効果
次に、共鳴効果も考えていきます。
孤立電子対を持つ置換基
例として、置換基をアルコキシ基だったときを考えます。
下図はオルト位に求電子剤が付加した際の中間体の共鳴構造ですが、孤立電子対があるため、共鳴構造を4つ書くことができます。
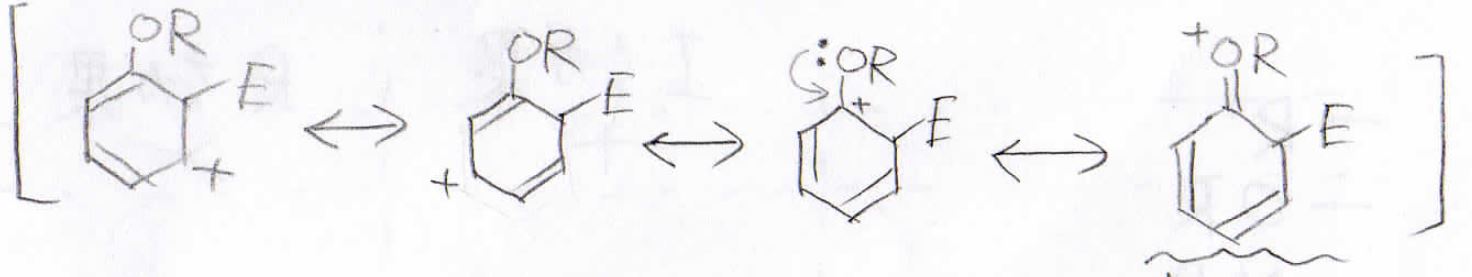
メタ位に付加した場合の共鳴構造は3つだけなので、オルト位に付加した場合のほうが安定な中間体であると言えます。
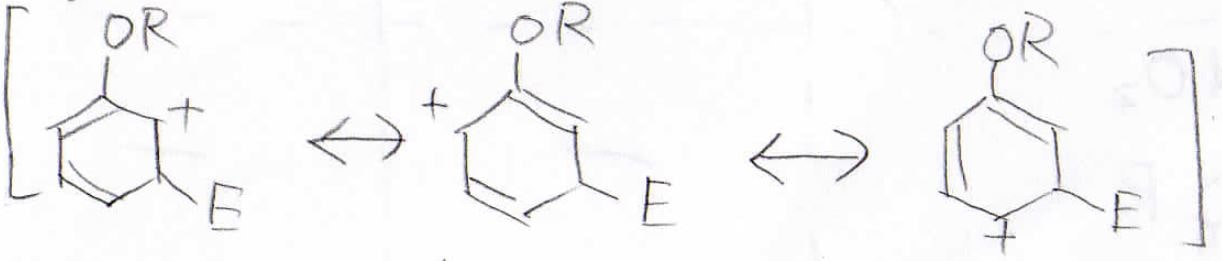
パラ位に付加した場合も、オルト位と同様に4つの共鳴構造を書くことができます。
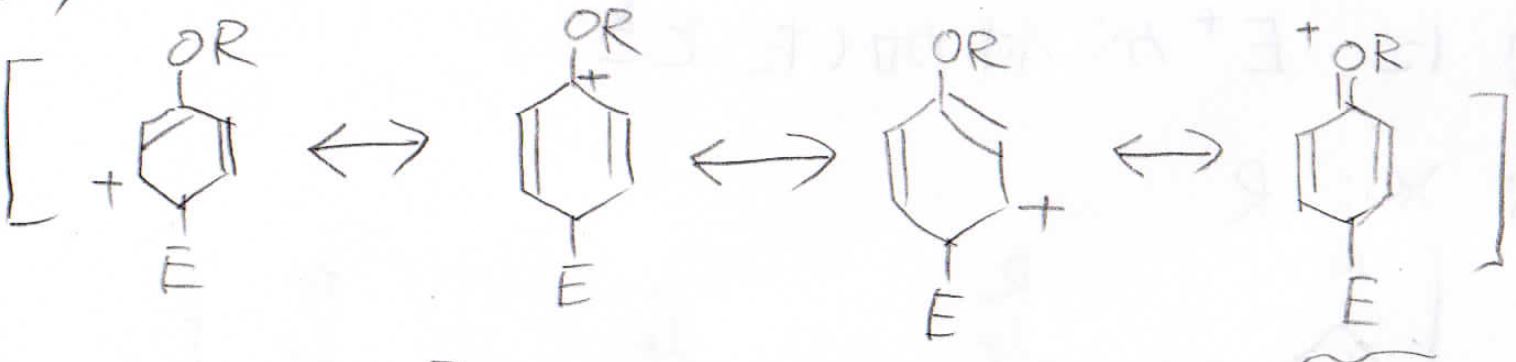
したがって、置換基に孤立電子対があった場合はオルト位かパラ位が置換されやすいということになります。
電子が吸い込める置換基
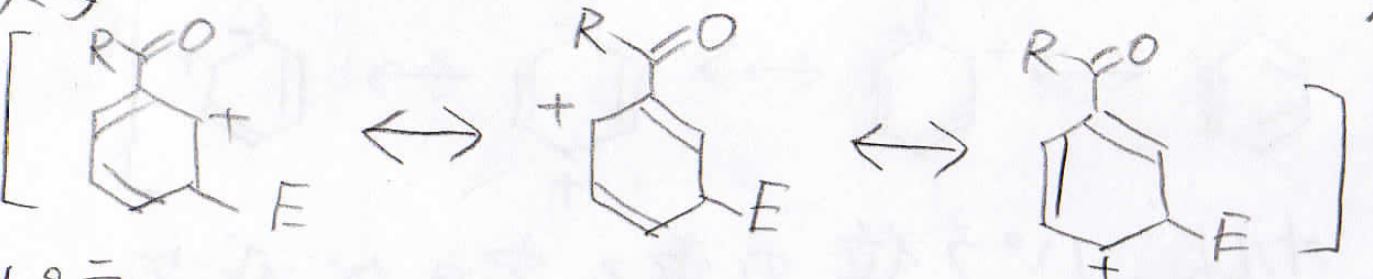
最後に、置換基がアシル基の場合も見てみましょう。
まず、オルト位に付加した場合ですが、酸素原子に電子を受け入れた結果、下図波線部のような共鳴構造の中間体ができます。
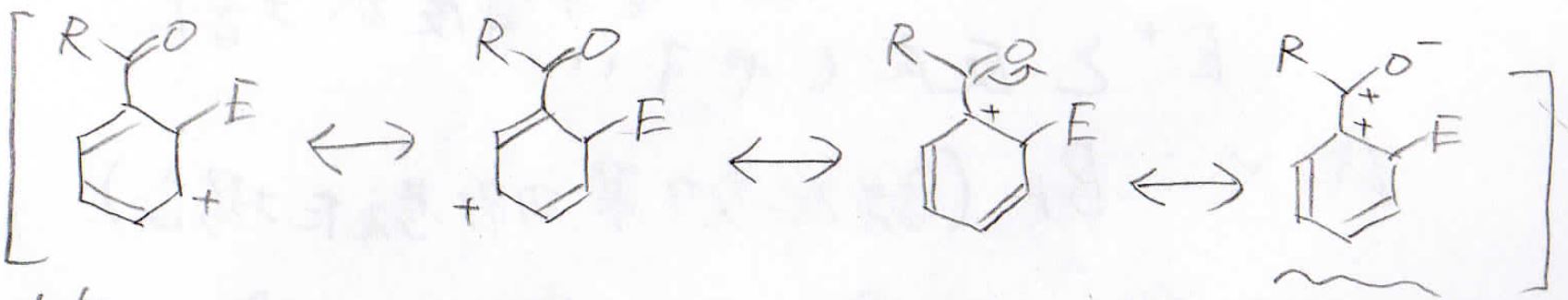
この構造は、正に帯電した原子が隣接しており、非常に不安定です。
この共鳴構造のおかげで、この中間体が不安定ということになります。
メタ位に付加した場合は、そのような不安定な共鳴構造は見られません。
パラ位に付加した場合はオルトと同じように不安定な共鳴構造が現れます。
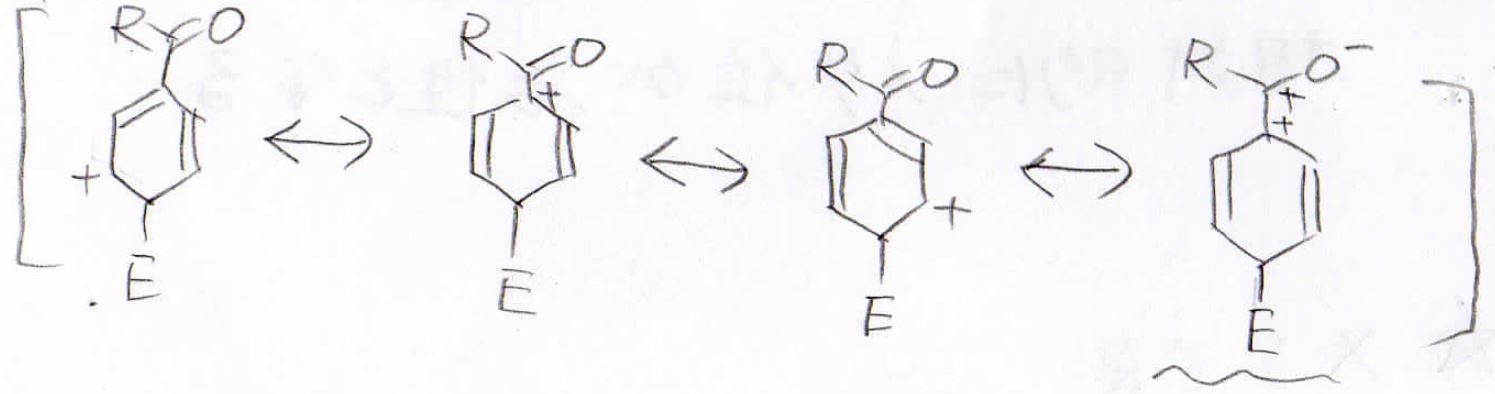
メタ位に付加した中間体が相対的に最も安定になるので、この場合はメタ置換体ができやすいということになります。
各置換基の配向性と活性化度
以上のことをまとめると、次のとおりになります。
配向性

まず、アルキル基は共鳴効果がないので、誘起効果のみで配向性が決まって、オルト-パラ配向性になります。
酸素原子や窒素原子がベンゼンに結合した構造は誘起効果、共鳴効果いずれもはたらきますが、この場合は共鳴効果の方が優先されてオルト-パラ配向性になります。
ハロゲンも電気陰性度で考えれば電子求引基ですが、孤立電子対があるので、オルト-パラ配向性になります。
ニトロ基やアシル基では電子を受け入れられる構造があるため、オルト、パラ置換体が相対的にできにくくなり、メタ配向性となります。
ただし、本当にメタ位の反応性が上がったわけではなく、あくまで相対的に反応性が高いという話ですので、反応速度は遅いです。
活性化度
最後に、求電子置換反応の活性化度について考えます。
これは、ベンゼン環内の電子密度で決定します。
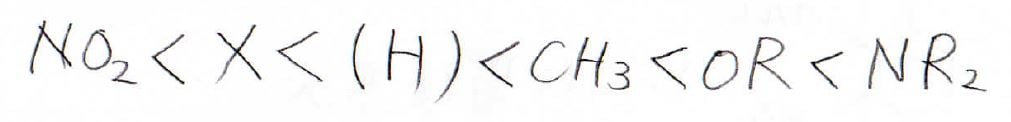
ニトロ基やアシル基は、ベンゼン環内の電子密度を大きく下げるので、最も反応しにくい部類に入ります。
ハロゲンは配向性こそ共鳴効果で決まりますが、電子密度においては電気陰性度の効果が大きいので、ベンゼン環内の電子密度は減少します。
したがって、活性化度はベンゼンより低くなります。
アルキル基は共鳴効果がない分、少し活性なぐらいです。
アルコキシ基やアルキルニトロ基では、共鳴効果により活性化度が大きく増大します。
アニリンやフェノールでも、求電子置換反応は起こりやすいです。
二置換体以上の配向性
ここからは、すでに2つ置換基がついているベンゼンにさらに求電子置換反応を起こした場合、どこが置換されやすいのか、考えていきます。
競合しない場合
例えば、p-メトキシベンゼンスルホン酸について、3つ目の置換基がつく部位は、図の中でAとBの2か所が考えられます。
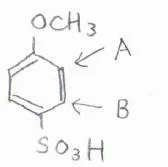
どちらが置換されやすいのか考えるには、まずそれぞれの置換基単体での配向性を考えることが有効です。
メトキシ基はオルト-パラ配向性であるため、オルト位にあたるAの位置のほうが置換されやすいです。
スルホン基はメタ配向性であり、メタ位にあたるのはAの位置になります。
どちらの置換基からみてもAのほうが置換されやすいと予測でき、実際のp-メトキシベンゼンスルホン酸でも、そのようになります。
同様に、こちらに挙げた二置換体では、2つの置換基の配向性が競合せず、矢印で示した位置が置換されやすいと言えます。
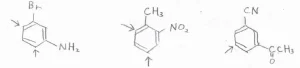
競合する場合
それぞれの配向性を考えたときに、異なる位置が置換されやすいことになる場合は、実験的に決められた配向性の強さによって、一方の置換基の配向性を優先させます。
その配向性の強さの順番は、こちらのようになります。

アミノ基、ヒドロキシ基、アルコキシ基など、求電子置換反応を大きく活性化させる置換基ほど優先されます。
そして、アルキル基は少し活性化、ハロゲン置換基は少し不活性化させる置換基ですが、同程度に優先されるものと考えてください。
大きな不活性化が起こるメタ配向性の置換基は、優先順位が低いです。
ここでは、例として3つの化合物を挙げています。
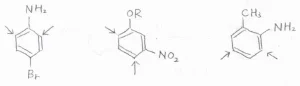
左のp-ブロモアニリンでは、アミノ基とブロモ基はどちらもオルト-パラ配向性ですが、アミノ基が優先されるので、矢印で示した位置で置換が起こりやすいということになります。
中央のm-アルコキシニトロベンゼンでは、オルト-パラ配向性のアルコキシ基が優先されます。
アルコキシ基とニトロ基の間にあたる位置では立体反発が大きいので置換は起こりにくく、結局、矢印で示した2か所が置換されやすい位置となります。
右のo-メチルアニリン(慣用名: o-トルイジン)でも、アミノ基が優先されて、図の中の2か所が置換されやすくなります。
そして、その下に書いた3つの化合物は、優先順位が同じ置換基どうしで競合が起こっているものです。

こういった場合は、基本的にはどちらの配向性も効いてくることにはなりますが、嵩高い置換基から見て、オルト位にあたる位置は、やはり置換されにくいです。
また、1,3-置換体における2位の位置、つまり2つの置換基の間の位置も置換されにくいです。
以上の法則は、三置換体以上でも同様に成り立ちます。
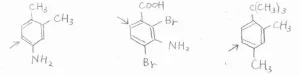
ここには、三置換体および四置換体で、もっとも置換されやすい位置を示しています。左と中央の化合物では、オルト-パラ配向性であるアミノ基が優先されます。
右の化合物では、立体反発が最も小さい位置で置換が起こります。
多置換ベンゼンの合成戦略
置換基の相互変換
ここからは、さらなる応用として、置換基を別の置換基へと変換させることで、配向性の制御をするテクニックについて見ていきます。
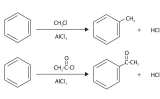
アルキル基とアシル基
例えば、アルキル基は酸化剤を加えてアシル基に変えることができますが、これにより、オルト-パラ配向性からメタ配向性へ変化します。
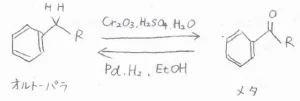
アルキル基のメタ位を狙って求電子置換反応させたい場合には、最初にアルキル基をアシル基に変換させて置換させたのちに、アシル基をアルキル基に戻すという経路を考えることができます。
アシル基をアルキル基に還元する反応には、パラジウム触媒共存下での水素化のほか、Clemmensen還元やWolf-Kishner還元という方法があります。
詳しくは、こちらの記事を参照してください。
アミノ基とニトロ基
続いて、アミノ基とニトロ基も互いに変換が可能な置換基です。

アミノ基は、トリフルオロ過酢酸を用いて酸化させることで、ニトロ基に変換できます。
この際、オルト-パラ配向性からメタ配向性へ変化します。
ニトロ基からアミノ基への還元は、鉄やスズ、亜鉛といった金属と塩酸を用いることで進行します。
スルホン基による保護と脱保護
また、違うパターンとして、スルホン基による保護というテクニックがあります。

例えば、tert-ブチルベンゼンを原料として、そのオルト位をニトロ化することを考えます。
tert-ブチル基は、オルト-パラ配向性ですが、立体反発が大きいため、そのままだと、その多くがパラ置換体になってしまいます。
そこで、始めにパラ位を発煙硫酸によりスルホン化させることで、パラ位を保護します。
このとき、2つの置換基の配向性は競合せず、次に置換されやすいのは、tert-ブチル基から見てオルト位になります。
硫酸と硝酸の混酸を用いてニトロ化した後に、希硫酸を用いて脱保護することで、o–tert-ブチルニトロベンゼンが得られます。
Friedel-Craftsアシル化の順序
合成戦略を立てるうえでは、配向性だけでなく、ベンゼン環内の電子密度にも注意が必要です。
特に、強く不活性化されたベンゼン環では、Friedel-Crafts反応を進行させるのが困難になるので、電子密度が大きいうちにやっておかなくてはいけません。
例えば、ベンゼンからm-ニトロアセトフェノンを作りたい場合、アセチル基をつけてから、ニトロ基をつけるというように順番があります。
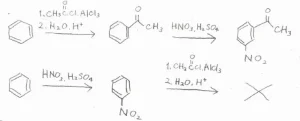
順番を逆にすると、ニトロベンゼンからのFriedel-Craftsアシル化がうまく進行しません。
アミノ基、ヒドロキシ基の保護
また、ベンゼン環が強く活性化されることで、うまく制御できなくなることもあります。
例えば、アニリンのオルト位かパラ位の1か所をブロモ化しようとしたとき、求電子置換が一気に起こることで、3か所がブロモ化された化合物ができます。
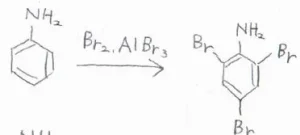
フェノールでも同様の現象が起こります。

さらに、アミノ基やヒドロキシ基のように孤立電子対をもつ置換基では、Lewis塩基として求電子剤に作用し、反応が複雑になる可能性があります。
アニリンに置換反応を1回だけ起こすためには、活性を下げるためにアセチル基で保護するというテクニックがあります。
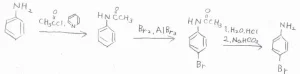
まず、触媒量のピリジンと塩化アセチルまたは無水酢酸を用いることで、アニリンからアセトアニリドを作ります。
これをブロモ化すると、電子密度の大きさと立体反発の大きさからパラ位が選択的に置換されます。
最後に、酸を加えて塩酸塩とした後、炭酸水素ナトリウムなどで中和すれば、アセチル基を外すことができます。
フェノールの場合は、ヨウ化メチルを用いてメトキシベンゼンとすることで、一置換までに制御できます。
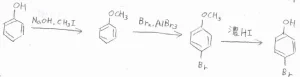
酸性条件下での加水分解によって、脱保護できます。
多環ベンゼン系炭化水素の配向性
ここからは、ナフタレンやアントラセンといった多環ベンゼン系炭化水素といわれる化合物の配向性について、考えていきます。
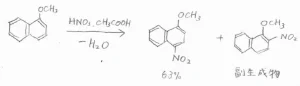
まず、ナフタレンは1位の炭素およびそれと等価な3つの炭素の位置が置換されやすいです。
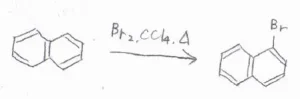
ナフタレンに臭素分子を反応させると、1-ブロモナフタレンができます。
Lewis酸触媒がなくても穏和な条件の下で進行することから、ナフタレンはベンゼンよりも求電子置換反応に対して活性であると言えます。
その位置選択性の理由は、反応機構から説明されます。
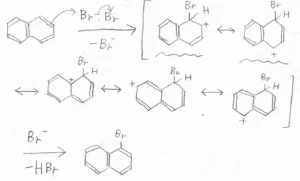
下図は、1位の炭素が攻撃されてできる中間体の共鳴構造です。
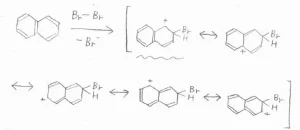
そして、仮に2位の炭素が攻撃されてできる中間体の共鳴構造は、以下のとおりです。
どちらも5つの共鳴構造があって、安定な中間体であるように見えますが、なぜ攻撃される位置に選択性があるのでしょうか?
ここで注目してほしいのは、波線を引いた3つの共鳴構造です。
これらは、電子が非局在化できるベンゼン環構造を保持したままのもので、特に安定な共鳴構造です。
前者のパターンでは、そんな構造が2つあるのに対して、後者のパターンでは1つしかないので、中間体の安定性に違いが生じ、より安定なC1攻撃が選択的に進行するということです。
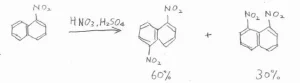
さらに、ナフタレンの一置換体に対して、さらに求電子置換反応を起こそうとした場合にも、位置選択性が現れます。
2つのベンゼン環のうち、置換基がある一方だけが強くその影響を受けるため、活性化基なら同じベンゼン環のオルト位(C2)やパラ位(C4)、不活性化基ならもう一方のベンゼン環のほうが活性なので、5位と8位が優先的に置換されます。
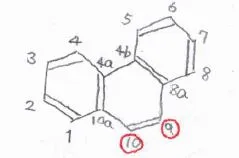
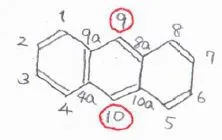
芳香環を3つもつ場合も考え方は同じで、フェナントレンとアントラセンは、いずれも9位と10位が最も置換されやすい位置になります。
練習問題
それでは最後に、練習問題をやってみましょう。
(1)tert-ブトキシベンゼンの配向性は?
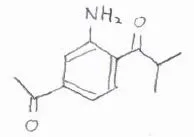
(2)下記の三置換体をベンゼンから合成する経路を考えてみてください。
すると、オルト-パラ配向性なので、メタ置換体ができにくいことがわかります。
ここで、tert-ブトキシ基の嵩高さを考慮すると、オルト位にはなかなか求電子剤が近寄りにくいということが予想されます。
したがって、tert-ブトキシベンゼンはオルト-パラ配向性ですが、オルト置換体よりもパラ置換体のほうが明らかに多くできやすいことになります。
(2)Friedel-Craftsアシル化は、不活性化されたベンゼン環では起こせないので、なるべく早い段階でやります。
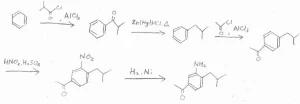

目的の化合物は、アシル基を2つもっているので、続けてFriedel-Craftsアシル化を2回やりたいところですが、アシル基は不活性化基かつメタ配向性なので、いったんアルキル基へ変換してから、2度目のアシル化をします。
そうしてできた二置換体では、配向性が競合しておらず、アルキル基から見てオルト位が置換されやすいので、ここをニトロ化して、アミノ基へ変換します。
最後に、酸化することでアルキル基をアシル基に変換すると、目的の構造となります。
また、別解として、アシル化、ニトロ化、アシル化という順番も考えてみました。

スルホン化と窒素のアセチル化を利用して、副生成物ができにくい経路を考えています。
最後のスルホン基の脱保護とN-アセチルアミノ基の加水分解は、どちらも酸性条件で起こせるので、同時に行うことができます。
まとめ
今回の内容は以上です。
間違いの指摘、リクエスト、質問等あれば、X(https://X.com/bakeneko_chem)かお問い合わせフォームよりコメントしてくださると、助かります。
それではどうもありがとうございました!